De Maroteaux-Lamy syndrom tillhör mukopolysackaridoserna, som innefattar olika lysosomala lagringssjukdomar. Syndromet beror på en genetisk mutation som resulterar i otillräcklig enzymaktivitet och leder till dermatosulfatavlagringar. Terapin består huvudsakligen av enzymersättningsterapier.
Vad är Maroteaux-Lamy syndrom?

© imaginuma - stock.adobe.com
Mukopolysackaridoserna är en oberoende grupp av sjukdomar som innehåller lysosomala lagringssjukdomar. Lysosomala lagringssjukdomar finns runt hörnet. Alla är genetiskt bestämda metabola sjukdomar som orsakas av funktionsfel i lysosomen. En av dessa sjukdomar är den så kallade Maroteaux-Lamy syndrom. Den medfödda metaboliska störningen leder till lagring av dermatinsulfater.
Termen används som synonymer Mukopolysackaridos typ VI, Arylsulfatas B-brist, ARSB-brist och ASB-brist Till exempel N-acetylgalaktosamin-4-sulfatasbrist. Sjukdomen beskrevs först 1963. De parisiska mänskliga genetikerna och barnläkarna P. Maroteaux och M. Lamy anses vara de första som beskriver den.
Förekomsten av sjukdomen är mellan en och nio drabbade av 100 000 personer. Familjeansamling observerades i de fall som hittills dokumenterats. Syndromet ärvs på ett autosomalt recessivt sätt. En genetisk mutation anses vara orsaken till sjukdomen.
orsaker
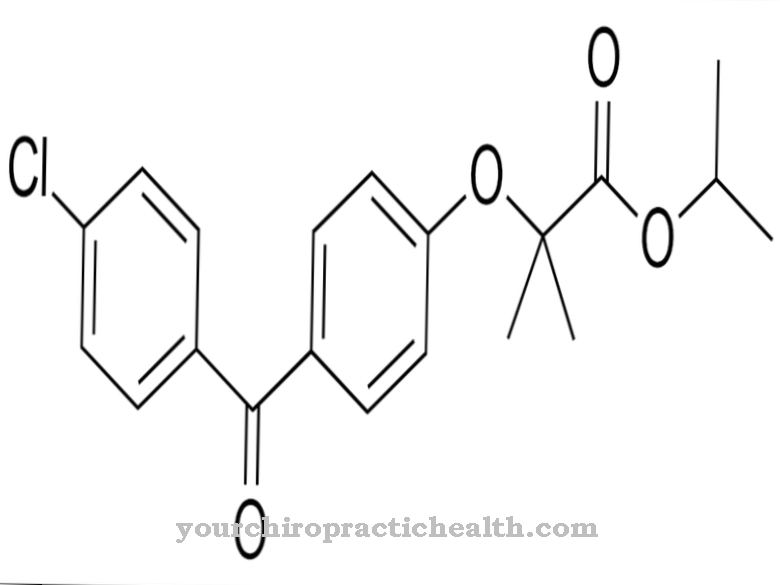
Maroteaux-Lamy syndrom beror på en mestadels ärftlig mutation. De orsakande mutationerna har nu lokaliserats till ARSB-genen. Mutationer vid genlokuset 5q13 till 5q14.1 sägs kunna orsaka symptomkomplexet. De gener som finns där kodar inom DNA för ett specifikt protein.
En mutation av generna sägs resultera i onormal aktivitet av det muterade arylsulfatas B, även känt som ASB eller N-acetylgalaktosamin-4-sulfatas. Ämnets aktivitet reduceras som en del av mutationen. På grund av denna minskning finns det störningar i nedbrytningen av ämnen som kondroitinsulfat och dermatansulfat. Eftersom ämnena inte längre bryts ned tillräckligt på grund av kausal mutation, lagrar kroppen rester av ämnena.
De typiska symtomen på Maroteaux-Lamy syndrom är ett resultat av denna lagring. Det är ännu inte känt om genetiska faktorer såväl som yttre påverkan spelar en roll i utvecklingen av mutationen. Detta kan antas, åtminstone för nya mutationer.
Symtom, åkommor och tecken
Patienter med Maroteaux-Lamy syndrom lider av ett komplex av kliniskt karakteristiska kriterier. Ett av de viktigaste symtomen är en oproportionerligt verkande kort statur, som kännetecknas av en kort bagageutrymme. Patientens oproportionalitet förknippas med ett grovt ansikte som påminner om symptomen på Hurlers sjukdom.
I de flesta fall är patientens hornhinna molnig. Dessutom kan det finnas hepatosplenomegals, hernias eller sammandragningar i lederna. Patienternas hjärtventiler blir allt mer tjockare på grund av avlagringar. Dessutom kan det finnas skelettdysplasi som liknar den hos patienter med dysostosmultiplex.
Både den kliniska bilden och förloppet av syndromet anses vara varierande och individuella. Förutom långsamma processer dokumenterades också snabba processer. Om de första symtomen visar sig omedelbart efter födseln talar detta fenomen för en ganska snabb kurs.
Detta gäller särskilt för en ökning av glykosaminoglykan i urinen, för svår dysostosmultiplex och uppvisar kort statur. Berörda personer med långsam kurs visar vanligtvis de första symptomen mycket senare. Ökningen av GAG är mycket lägre och dysostosmultiplexet är mycket mildare.
Diagnos & sjukdomsförlopp
Maroteaux-Lamy syndrom behöver inte nödvändigtvis manifestera sig omedelbart efter födseln och diagnostiseras i många fall först senare, när de första symtomen uppträder. Diagnosen är baserad på de kliniskt typiska kriterierna för sjukdomen och baseras därför främst på en signifikant reducerad ASB-aktivitet, som kan spåras på odlade fibroblaster och leukocyter.
Å andra sidan finns normal aktivitet i förhållande till andra sulfataser. Som en del av diagnostiken ger läkaren också bevis på den ökade mängden dermatansulfat som utsöndras i urinen. Vid differentierad diagnos måste Maroteaux-Lamy-syndrom differentieras från multipel sulfatasbrist och andra former av mukopolysackaridos. Sialidos och mucolipidosis är också sjukdomar som är relevanta för differentiell diagnos. Patientens prognos varierar från fall till fall och beror främst på början och hur allvarliga de första symptomen är.
komplikationer
Först och främst leder Maroteaux-Lamy-syndrom till en kort status hos patienten. Barn i synnerhet kan drabbas av mobbning och retad i ung ålder och utveckla psykologiska problem eller depression som ett resultat. Som regel sker inte ytterligare tillväxt av patienten proportionellt och olika klagomål och missbildningar inträffar, som också förekommer i ansiktet.
Dessutom är hjärtventilerna skadade och missplacerade av Maroteaux-Lamy-syndrom, så att det finns klagomål eller begränsningar i hjärtat. I vissa fall kan det också minska patientens livslängd, vilket kan leda till hjärtdöd. En kausal behandling av detta syndrom är inte möjlig. Av detta skäl är huvudsyftet med behandlingen att begränsa och bekämpa symtomen så att den drabbade kan leva ett vanligt liv.
Psykologisk behandling kan också vara nödvändig. Det finns inga speciella komplikationer, även om inte alla klagomål kan vara helt begränsade. Som regel reduceras inte förväntad livslängd med Maroteaux-Lamy-syndrom om det inte finns några symtom på hjärtat.
När ska du gå till läkaren?
Om ett växande barn visar tecken på störningar eller förändringar i den fysiska utvecklingsprocessen, måste en läkare konsulteras. Om barnet är tydligt kort eller har en missbildning av skelettet behöver han medicinsk hjälp. Om det finns onormala eller speciella egenskaper i kroppsformen i direkt jämförelse med barn i samma ålder, bör en läkare konsulteras för att klargöra symtomen. Störningar i rörelsesekvenser eller allmän motorisk färdighet måste presenteras för en läkare. En läkare kommer också att behövas om hornhinnan är grumlig eller synen minskar. Oregelbundna hjärtrytmer indikerar ett hälsoproblem, som måste undersökas i olika medicinska test.
I de flesta fall kan de första avvikelserna i sjukdomen upptäckas omedelbart efter födseln på grund av förändringar i skelett. Eftersom spädbarn granskas omfattande av läkaren när de föddes, finns det inget behov av föräldrarna att agera. Om symtom som kräkningar eller smärta uppträder under de första veckorna eller månaderna av livet, måste en läkare konsulteras. Om barnet gråter och gråter kontinuerligt är detta ett tecken på en oegentlighet som måste klargöras. Vid andfåddhet eller akut hälsotillstånd måste en larmtjänst varnas. Fram tills den anländer måste första hjälpen ges för att säkerställa barnets överlevnad.
Behandling och terapi
En kausal terapi är inte tillgänglig för patienter med Maroteaux-Lamy-syndrom i smalare bemärkelse, eftersom den förändrade enzymaktiviteten hos de drabbade beror på en genetisk mutation som endast kan åtgärdas genom genterapi. Men med enzymersättningsterapi i vidaste bemärkelse finns en typ av terapi tillgänglig som adresserar symtomen på sjukdomen vid deras källa.
Vid enzymersättningsterapi får patienter galsulfas i betydelse av naglazym. Denna enzymersättning resulterar i en bättre uppdelning av de relevanta substanserna och försenar således sjukdomsförloppet. Emellertid kan de symptom som hittills uppstått inte fullständigt vändas. Förtjockning av hjärtventiler kan behandlas symptomatiskt och kan under vissa omständigheter kräva kirurgisk utbyte av hjärtventilerna.
Akuta hernias behandlas med taxibilar. Målet är en minskning som ger läkaren tid att hitta en operativ lösning. Allvarliga korneala opaciteter som resulterar i blindhet kan möjligen vändas med en hornhinnetransplantation. Symtom som kort statur kan i slutändan inte vändas, men förekommer ofta endast i mild form med tidig behandling med ett enzymersättande läkemedel.
Du hittar din medicin här
➔ Läkemedel mot smärtaOutlook & prognos
Denna sällsynta sjukdom förekommer i en mängd olika former och former.Tack vare den individuella kursen och de olika graderna av Maroteaux-Lamy-syndrom eller mukopolysackaridos typ 6 är det vanligtvis svårt att ge en tillförlitlig prognos för en specifik patient.
Generellt sett är prognosen sämre om de första symtomen på Maroteaux-Lamy syndrom dyker upp strax efter födseln. Detta talar vanligtvis för en snabbare sjukdomsförlopp.
Som ett resultat kan man säga att åldern för de drabbade gör det möjligt att dra en slutsats om den förväntade prognosen, liksom tidpunkten då de första symptomen på Maroteaux-Lamy-syndrom dök upp. Dessutom beror utsikterna för patienten också på kvaliteten på behandlingen. Den tidpunkt då enzymersättningsterapi inleddes är ofta kritisk.
Enzymersättningsterapi kan bryta ner ämnen som kondroitinsulfat och dermatansulfat. Det bromsar således sjukdomsförloppet. Problemet är dock att skador som redan har inträffat i organismen vanligtvis inte är reversibla. Detta minskar livskvaliteten, men kan också leda till att den drabbade personen dör tidigare. Förloppet av Maroteaux-Lamy syndrom kan vara snabbt om det inträffar tidigt. Patienten kan också svara bra på den medicinering som administreras. I detta fall kommer Maroteaux-Lamy-syndromet att utvecklas långsamt.
förebyggande
Hittills är inga externa faktorer kända för utvecklingen av Maroteaux-Lamy syndrom. Den enda förebyggande åtgärden för närvarande är genetisk rådgivning i familjeplanering.
Eftervård
Eftersom behandlingen av Maroteaux-Lamy-syndrom är komplex och på lång sikt krävs ingen klassisk uppföljning. Snarare bör de drabbade koncentrera sig på att hantera sjukdomen säkert och att bygga en positiv inställning trots motgången. Avkopplingsövningar och meditation kan också hjälpa till att lugna och fokusera sinnet. Eftersom kort statur går hand i hand med en minskning av estetik, bör eventuella underlägsenhetskomplex och svag självkänsla diskuteras med en terapeut vid behov. Detta kan bidra till att bättre acceptera sjukdomen och förbättra livskvaliteten på lång sikt.
Du kan göra det själv
Alternativ för självhjälp är inte tillgängliga för dem som drabbats av Maroteaux-Lamy syndrom. Sjukdomen kan endast behandlas symptomatiskt; det finns ingen kausal terapi.
För att lindra symtomen på sjukdomen beror de drabbade på intaget av enzymer och olika mediciner. Här bör regelbundet och föreskrivet intag säkerställas. I svåra fall är emellertid kirurgiska ingrepp i hjärtat nödvändiga. För att inte stressa hjärtat i onödan bör onödig ansträngning undvikas. Detta gäller särskilt för plötsliga eller plötsliga belastningar.
Om patienten eller föräldrarna vill få barn igen kan genetisk rådgivning vara användbar för att förhindra att Maroteaux-Lamy-syndrom uppstår igen. Samtal med nära människor eller vänner kan ofta lindra psykologiska klagomål eller depression. Kontakt med andra patienter med Maroteaux-Lamy-syndrom har ofta en mycket positiv effekt på sjukdomen och kan bidra till ett informationsutbyte som möjligen kan förbättra livskvaliteten för den drabbade. Emellertid kan inte fullständigt botande av syndromet uppnås.
.jpg)









.jpg)


.jpg)