De Jag cellsjukdom är en lyosomal mukolipidos. Orsaken till lagringssjukdomen är en mutation av GNPTA-genen med gen locus q23.3 på kromosom 12. Symtomatisk behandling utförs huvudsakligen genom administrering av bisfosfonater.
Vad är jag cellsjukdom?

© red150770 - stock.adobe.com
Lagringssjukdomar kännetecknas av avsättning av olika ämnen i celler och organ i människokroppen. Det är en heterogen grupp sjukdomar som kan delas upp i flera underformer. Förutom glykogenoser, mukopolysackaridoser och lipidoser, skiljer medicinen sig beroende på det avsatta ämnet, sfingolipidoser, hemosideroser och amyloidoser.
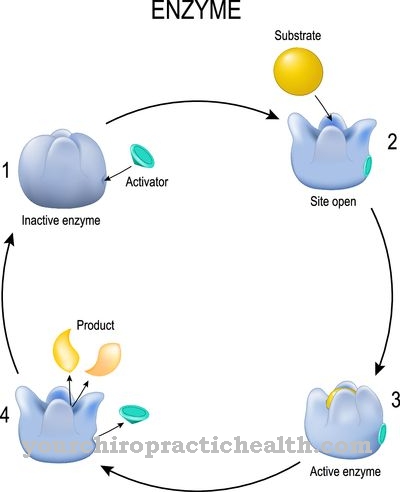
Lysosomala lagringssjukdomar påverkar lysosomerna. Dessa är små membranbelagda cellorganeller i eukaryoterna. Lysosomer bildas av Golgi-apparaten och är utrustade med hydrolytiska enzymer och fosfataser. Med hjälp av sina enzymer bör de främst smälta främmande ämnen och kroppens egna ämnen.
I-cellsjukdom är en lyosomal mukolipidos med två olika subtyper. Leroy och DeMars dokumenterade först sjukdomen på 1960-talet och pekade på dess likhet med mukopolysackaridos typ I, känd som Hurlers sjukdom. Sjukdomens namn kommer från fibroblastinklusioner, de så kallade inklusionscellerna, i patientens hud.
orsaker
Orsaken till I-cellsjukdomen ligger i en brist på aktivitet av N-acetylglukosaminyl-1-fosfotransferas. Den begränsade aktiviteten hos detta enzym förhindrar att en stor del av de lysosomala enzymerna kommer in i lysosomets inre. Regleringen av de lysosomala enzymerna formas av aktiviteten hos fosfotransferas.
Det möjliggör syntes av en sorteringssignal i en frisk organisme. Denna process störs vid I-cellsjukdomar. Därför finns det ingen märkning med mannos-6-fosfat. Av detta skäl sorteras de lysosomala enzymerna inte längre korrekt och migrerar in i den extracellulära matrisen på ett okontrollerat sätt via plasmamembranet.
Anledningen till detta är en mutation i GNPTAB-genen. Det tar bort funktionaliteten hos N-acetylglukosaminyl-1-fosfotransferas och därmed förmågan att katalysera syntesen av mannos-6-fosfat. Transporten av lysomala enzymer är så störd. N-acetyl-glukosamin-1-fosfotransferaset består av underenheterna alfa, beta och gamma. De är kodade på två gener.
Den ärftliga I-cellsjukdomen påverkar GNPTA-genen på kromosom 12. Det finns en mutation i q23.3-genen. För den sällsynta sjukdomen ges en förekomst av cirka 0,3: 100 000. Arv är föremål för autosomal recessiv arv. Båda föräldrarna måste därför bära den defekta genen för att överföra sjukdomen.
Symtom, åkommor och tecken
I de flesta fall kan symtomen på I-cellsjukdomar observeras omedelbart efter födseln eller några månader senare, och deras egenskaper liknar Hurls syndrom. Till skillnad från patienter med Hurlers syndrom uppvisar de med I-cellsjukdom ingen utsöndring av mukopolysackarid.
De enskilda symtomen på sjukdomen utsätts för ett stort antal variationer. Kornfeld och Sly sammanfattar kliniska egenskaper hos skelettet, inre organ, ögon, hud, centrala nervsystemet och ansiktet. Skelettet påverkas så ofta av kyfoskolios och hoftdislokationer.
Klubbfotar, ledkontrakturer och deformiteter i ryggkotorna kan också vara närvarande. Detsamma gäller för kortstatus och dysostosmultiplex. Sjukdomen kan manifestera sig i de inre organen i form av hepatosplenomegaly och kardiomegali eller hjärtsjukdom. Patientens ansikte har grova drag.
Exofthalmos, hyperplastiskt tandkött eller scaphocephaly är typiska symtom. Också kännetecknande är en öppen mun och en djupt nedsänkt näsa. De drabbade ögonen har ofta korneala opaciteter eller svullna ögonlock. Huden är tjock och grov, med svår psykomotorisk eller mental retardering i centrala nervsystemet.
Diagnos & sjukdomsförlopp
Den första misstänkta diagnosen I-cellsjukdom kan göras genom visuell diagnos baserad på anamnesen. En biokemisk bestämning av den lysosomala enzymaktiviteten i serumet kan användas för att bekräfta diagnosen. Denna bestämning avslöjar ett absurt samband mellan intra- och extracellulär aktivitet.
Aktiviteten av fosfotransferas i fibroblasterna kan också bestämmas för att bekräfta diagnosen. Inneslutningarna motsvarar antingen mukopolysackarider, lipider eller oligosackarider. Molekylär genetisk diagnostik kan skingra alla kvarvarande tvivel. Om det finns en lämplig historia kan sjukdomen också diagnostiseras som en del av prenatal diagnos.
På grund av den låga prevalensen rekommenderas egentligen prenatal upparbetning endast om det finns en familjedisposition. Sjukdomsförloppet beror på symtomen i det enskilda fallet och är inte direkt förutsägbart. De flesta patienter överlever dock knappt tio års ålder. Mildare utvecklingsformer är dock inte helt uteslutna i enskilda fall.
komplikationer
I-cellsjukdom kan leda till olika komplikationer och klagomål. Dessa erkänns dock sent, så att I-cellsjukdomen endast kan diagnostiseras sent. Symtomen är relativt inkonsekventa, vilket ofta gör behandlingen svår. Detta leder vanligtvis till obehag och missbildningar i hud, ögon och inre organ.
I värsta fall kan den drabbade blinda eller dö direkt från organsvikt. Dessutom finns det en uttalad kort statur och hjärtproblem. Ögonlocken är ofta svullna och det är minskad intelligens och mental retardering. Det är inte ovanligt att den drabbade är beroende av hjälp av andra människor i vardagen på grund av retardationen för att hantera det.
Patientens livskvalitet reduceras kraftigt av I-cellsjukdomen. Som regel finns det inga speciella komplikationer vid behandling av sjukdomen. Läkemedel och psykologiska behandlingar används som kan lindra symtomen. Emellertid är en komplett och kausal behandling av denna sjukdom inte möjlig. Livslängden minskas av sjukdomen.
När ska du gå till läkaren?
I-cellsjukdom diagnostiseras vanligtvis omedelbart efter att barnet föddes. Huruvida ytterligare behandlingsåtgärder är nödvändiga beror på symtomens typ och svårighetsgrad. Mindre missbildningar behöver inte nödvändigtvis behandlas. Klubbfotar och deformiteter i ryggkotorna är å andra sidan allvarliga missbildningar som måste behandlas kirurgiskt och med medicinering. Föräldrar bör omedelbart konsultera en specialist om den läkare som är ansvarig på modersjukhuset inte redan har gjort det.
Om en olycka eller fall inträffar till följd av klagomålen måste barnet föras till sjukhus eller föräldrarna ska omedelbart ringa räddningstjänsten. Vid svåra missbildningar, som också kan påverka barnets psyke senare i livet, bör en terapeut konsulteras för att följa den medicinska behandlingen. I-cellsjukdomen kräver därför alltid en medicinsk undersökning. Rätt kontaktperson är barnläkare eller specialist på ärftliga sjukdomar. Vid synstörningar bör en ögonläkare konsulteras.
Läkare & terapeuter i ditt område
Behandling och terapi
I-cellsjukdomen anses obotlig. En kausal terapi finns därför inte. Behandlingen är endast symtomatisk och stödjande. Psykoterapeutisk vård för drabbade familjer utgör en stor del av stödjande terapi. Symtomatisk behandling beror på det enskilda fallet. Bensymptomen behandlas ofta genom att ge bisfosfonater.
Dessa läkemedel är kända från behandling av osteoporos och har en hög affinitet för benytan. Speciellt i regionen för resorptionslackorna fäster de sig vid benen. På så sätt hämmar de de bennedbrytande osteoklasterna och på detta sätt minskar benresorptionen. Läkemedlen är pyrofosfatanaloger med en kolinnehållande P-O-P-bindning.
En enzymatisk hydrolys äger inte rum på dem. Aminobisfosfonaterna är bland de senaste av dessa ämnen. Dessutom godkänns alendronat, klodronat, etidronat, ibandronat, pamidronat och risendronat i Tyskland från samma läkemedelsgrupp. Detsamma gäller för tiludronat och zoledronat.
Förutom dessa läkemedel kan benmärgstransplantationer också användas för att behandla I-cellsjukdomar. Framgången med denna behandling har endast varit begränsad i tidigare fall. Genterapier undersöks nu som en ny terapeutisk strategi för genfel. Genterapier har visat initial framgång i djurmodeller. Hittills har de inte kunnat användas i praktiken på människor. Men detta förhållande kommer förmodligen att förändras i framtiden.
Du hittar din medicin här
➔ Läkemedel mot smärtaOutlook & prognos
I-cellsjukdom är en ärftlig sjukdom som ännu inte har behandlats symptomatiskt. Prognosen är följaktligen negativ. Även om symptomen kan minskas betydligt genom tidig behandling, tar I-cellsjukdomen nästan alltid en allvarlig kurs.
Den korta staturen och skadorna på de inre organen och huvudet minskar redan livslängden avsevärt. Vidare kan missbildningar i ansikte, hud och ögon minska livslängden, men främst också livskvaliteten för den drabbade. Några av de drabbade når 40 eller 50 år, men de flesta dör i barndom eller ungdom.
Om I-cellsjukdomen inte behandlas, dör de sjuka ofta under de första åren av livet. Prognosen är därför ganska negativ. Icke desto mindre ges möjligheterna till ett relativt symptomfritt liv om patienten behandlas som en del av en omfattande terapi och vid behov placeras i en institution för fysiskt funktionshindrade. Fysioterapi och terapeutiska åtgärder kan förbättra patientens välbefinnande på lång sikt.
förebyggande
I-cellsjukdom kan endast förebyggas genom ett molekylärt genetiskt test innan familjeplanering. Som en del av den prenatala diagnosen kan förväntade föräldrar också besluta att avbryta graviditeten.
Eftervård
I de flesta fall har de som drabbats av I-cellsjukdom inga eller mycket få uppföljningsåtgärder tillgängliga. Sjukdomen måste erkännas av en läkare så tidigt som möjligt så att ytterligare försämring av symtomen kan förhindras. Eftersom detta är en genetiskt bestämd sjukdom, bör en genetisk undersökning och konsultation alltid genomföras först i fallet med en önskan att få barn för att undvika arv av I-cellsjukdomen till efterkommande.
De flesta patienter är beroende av olika mediciner för denna sjukdom. Det är viktigt att se till att doseringen är korrekt och att medicinen tas regelbundet. Om något är oklart, det finns biverkningar eller om du har några frågor, bör en läkare alltid konsulteras först.
På samma sätt behöver många drabbade psykologiska stöd för denna sjukdom, även om kärleksfulla diskussioner med föräldrar eller släktingar kan ha en positiv effekt på sjukdomsförloppet. En drabbad person behöver hjälp och stöd i vardagen från sin egen familj. I många fall begränsar eller reducerar I-cellsjukdomen den berörda personens livslängd avsevärt.
Du kan göra det själv
Patienter som lider av I-cellsjukdom kan använda olika konservativa och alternativa behandlingsmetoder. Konservativ terapi fokuserar på att lindra symtom och sjukdomar.
Användningen av hjälpmedel som kryckor eller ortopediska inläggssulor kan bromsa de respektive missbildningarna och därmed också minska smärtan. Medicinering hjälper till att lindra smärta och kan kompletteras med alternativa åtgärder som massage eller akupunktur. Alternativ behandling bör diskuteras med den ansvariga läkaren i förväg. Läkaren kanske kan hänvisa patienten direkt till en homeopat eller ge ytterligare tips om hur man behandlar respektive symptom.
Eftersom I-cellsjukdomen vanligtvis är dödlig trots alla behandlingsalternativ, bör terapeutiska råd sökas. Inte bara de drabbade bör arbeta genom sin rädsla. Släktingar och vänner behöver vanligtvis också stöd för att hantera sjukdomen och dess eventuella negativa resultat. Deltagande i en självhjälpsgrupp är också ett alternativ för patienten och deras anhöriga. Kontakt med andra drabbade hjälper till att acceptera sjukdomen, och ofta kan även andra drabbas av ytterligare behandlingsåtgärder och strategier för att leva dagligen med I-cellsjukdom.







.jpg)


.jpg)