De Allan-Herndon-Dudley-syndrom är en mutation i SLC16A2-genen som ändrar sköldkörtelhormontransportören MCT8 och orsakar försämrat jodtyroninupptag i muskelvävnad och i centrala nervsystemet. På grund av mutationen lider de drabbade av muskelsvaghet samt förseningar i mobil och mental utveckling. AHDS är obotligt och har hittills endast behandlats med administrering av triiodotyroacetat.
Vad är Allan - Herndon - Dudley syndrom?

Förseningar i den fysiska, mentala eller emotionella utvecklingen hos ungdomar och barn sammanfattas som utvecklingsförseningar eller retardering. Förseningar i utvecklingen kan ha olika orsaker. Exempelvis kan utlösaren för den försenade utvecklingen ligga i det centrala nervsystemet.
Detta är till exempel fallet med Allan - Herndon - Dudley Syndrome (AHDS). Förutom svår psykisk retardering kännetecknas syndromet av störningar i motorisk utveckling. Den första beskrivningen av syndromet går tillbaka till 1944. Den kliniska bilden du beskriver är en ärftlig sjukdom, dvs. en genetisk störning.
Sjukdomen drabbar manliga spädbarn i de flesta fall som hittills dokumenterats. Utvecklingsstörningar och deras konsekvenser är i nästan alla fall tydligt synliga från födseln. AHDS är en extremt sällsynt sjukdom.Av denna anledning har tillståndet till Allan - Herndon - Dudley syndrom hittills varit ganska dåligt.
orsaker
AHDS är en ärftlig genetisk störning orsakad av en mutation i SLC16A2-genen. Detta är den kodande genen för den så kallade sköldkörtelhormontransportören MCT8. Denna transportör förmedlar upptag av jodtyroniner i muskel- och nervvävnad.
På grund av mutationen uppstår störningar i upptaget av sköldkörtelhormoner, vilket upprör centrala nervsystemet och därmed försämrar utvecklingen av nervsystemets celler. Muskelvävnad och hjärna blir fattiga på grund av den mutationsrelaterade dysreguleringen av det aktiva sköldkörtelhormonet som de faktiskt är beroende av.
Syndromet överförs som en X-kopplad recessiv arv. Kvinnor kan ärva sjukdomen, men på grund av sin dubbla X-kromosomstruktur blir de sällan sjuka. Sjuka män kan inte reproducera sig.
Även om mutationen är genetisk, förutom denna interna faktor, spelar förmodligen externa faktorer en roll i sjukdomens början. På grund av sällsyntheten och den begränsade forskningsbasen har dessa yttre faktorer ännu inte slutgiltigt klargjorts.
Du hittar din medicin här
➔ Läkemedel mot muskelsvaghetSymtom, åkommor och tecken
Allan-Herndon-Dudley-syndrom är en medfödd sjukdom som vanligtvis manifesteras i småbarn eller spädbarn. De drabbade lider av mer eller mindre allvarlig muskelsvaghet. Barnens muskelvävnad är märkbart underutvecklad. Muskels svaghet åtföljs snart av leddeformiteter.
Kontraktioner är också ofta åtföljande symtom. Barnens rörlighet försämras alltmer av kontrakturer och deformiteter. Av denna anledning verkar de drabbade ofta onaturligt statiska eller till och med rörliga. På grund av det mutationsrelaterade underutbudet av sköldkörtelhormoner lider de drabbade ofta också av muskelkramper eller gör ofrivilliga rörelser med armar och ben.
Ofta kan de drabbade inte röra sig självständigt. I de flesta fall förknippas de motoriska nedsättningarna med allvarliga psykiska störningar. Till exempel kan majoriteten av patienterna inte tala. I enskilda fall kan AHDS kännetecknas av många andra symtom inom mental och fysisk utveckling.
Diagnos & kurs
Läkaren misstänker först AHDS i anamnesis. I laboratorietester indikerar en ökad T3-nivå med normala FT4- och TSH-nivåer Allan - Herndon - Dudley syndrom. Avbildning av det centrala nervsystemet är vanligtvis en del av diagnosen.
Muskelsvagheter på grund av motoriska neuronala sjukdomar kan uteslutas från den differentiella diagnosen. Prognosen för patienter med Allan-Herndon-Dudley-syndrom är relativt dålig. Hittills är sjukdomen obotlig. Studier har föreslagit att tidpunkten för diagnosen kan spela en avgörande roll i patientens prognos.
komplikationer
Liksom alla kromosonellt ärvda störningar kan inte Allan - Herndon - Dudley syndrom behandlas kurativt. Den vanligaste biverkningen av Allan - Herndon - Dudley syndrom - uttalad muskelsvaghet - kan behandlas med fysioterapi. En sådan behandling som syftar till att stärka musklerna kan vara smärtsam för patienten.
Särskilt små barn vägrar ofta terapi på grund av smärtan. Trots intensiv träning leder fysioterapi inte alltid till önskad framgång. Det liknar med talterapistödet från Allan - Herndon - Dudley-patienten. Även om minskningen av språkförmågan kan förbättras med intensiv träning, leder behandlingen inte alltid till framgång på grund av den mest högkvalitativa psykiska nedsättningen hos de drabbade.
Frustration från patientens sida och en stor börda för hela familjen är en av de allvarligaste komplikationerna i behandlingen av Allan - Herndon - Dudley syndrom. Muskelkramper och rörelser i extremiteterna som inte kan påverkas kan behandlas med administrering av muskelavslappnande medel. Komplikationer kan ses i de ibland allvarliga biverkningarna av läkemedlen.
Trötthet, en generell känsla av utmattning och malaise ska nämnas utöver stress på mag-tarmkanalen. Långvarig användning av relanxanter skadar också levern och njurarna. Om Allan - Herndon - Dudley syndrom inte behandlas, kommer de drabbade inte att kunna göra några väsentliga framsteg när det gäller deras mentala eller motoriska färdigheter.
När ska du gå till läkaren?
I många fall är direkt behandling av Allan-Herndon-Dudley syndrom inte möjlig. Av denna anledning är behandlingen huvudsakligen symptomatisk och riktar sig till individuella klagomål och förseningar. Som regel bör föräldrar då se en läkare om barnet har muskelsvaghet.
Detta kan märkas genom utmattning eller ihållande trötthet. Medicinsk rådgivning är också nödvändig om Allan-Herndon-Dudley syndrom bromsar mental och motorisk utveckling.
Om behandlingen inte äger rum under barndomen kan det leda till betydande symtom och begränsningar i vuxen ålder. En läkare bör konsulteras, särskilt om patienten inte längre kan tala. Behandling är också nödvändig för muskelkramper. Om det är akut akut kan du gå direkt till sjukhuset eller ringa ambulans.
I de flesta fall kommer Allan-Herndon-Dudley-syndrom att behandlas av en allmänläkare eller av en barnläkare. De enskilda klagomålen måste dock undersökas och behandlas av respektive specialist eller terapeut.
Läkare & terapeuter i ditt område
Behandling och terapi
AHDS är en orsakligt obehandlingsbar sjukdom. Eftersom det inte finns några terapier tillgängliga för att korrigera den primära orsaken har sjukdomen hittills inte varit härdbar. Under tiden antyder framsteg inom genterapi att genterapimetoder snart kommer att godkännas för klinisk vardag.
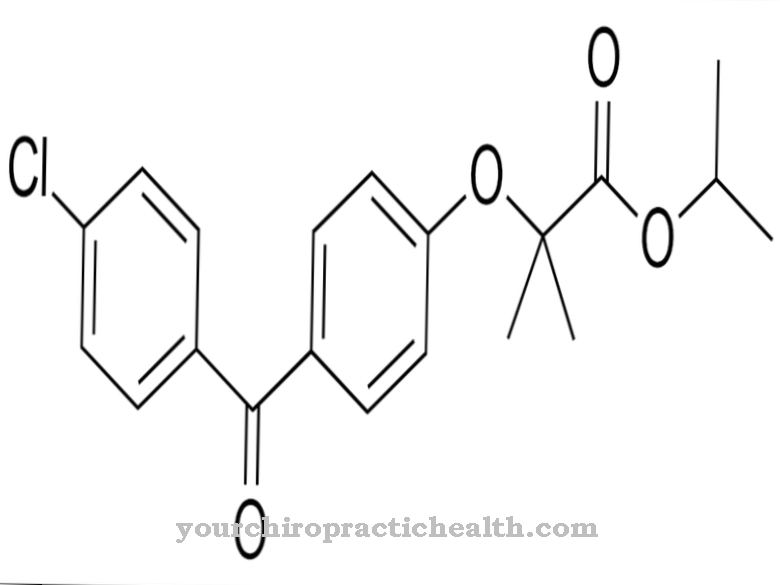
I vilken utsträckning patienter med syndromet skulle dra nytta av godkännande har ännu inte klargjorts. För närvarande finns det inget etablerat eller standardiserat behandlingsalternativ för patienter med AHDS inom området för symptomatisk behandling. För några år sedan ansåg forskare administrationen av TRIAC som ett möjligt symtomatiskt behandlingsalternativ.
TRIAC är ett icke-klassiskt sköldkörtelhormon, triiodotyroacetat. Administreringen av hormonet genomfördes i en klinisk studie på drabbade barn, men kunde inte uppnå några synliga resultat. Resultaten från studien är inte nödvändigtvis meningsfulla eftersom administreringen av hormonet började relativt sent.
Av detta skäl ansågs TRIAC 2014 fortfarande vara den bästa möjliga behandlingen. I ett fall under 2014 dokumenterades en betydande förbättring av motorisk och mental utveckling under behandling med TRIAC. Terapin startades på den drabbade personen i tidig barndom.
Resultaten av studierna hittills indikerar således att den tidpunkt då behandlingen påbörjas har en effekt på terapiresultaten som inte bör underskattas för patienter med AHDS. Ledsagande stödjande terapier som arbetsterapi och fysioterapi eller tidig intervention kan teoretiskt användas för att förbättra livskvaliteten och patienternas färdigheter. Det finns emellertid knappast några bevis på effektiviteten hos en sådan metod i samband med AHDS-patienter.
Outlook & prognos
Allan-Herndon-Dudley-syndrom orsakar ett antal olika symtom hos de flesta patienter. Först och främst drabbas de drabbade av svår muskelsvaghet. Detta innebär att normala aktiviteter eller idrott inte längre kan utföras för den berörda personen. Det finns också stora förseningar i intellektuell och mobil utveckling. Patientens koncentration är tydligt begränsad och minskad.
Det finns fortfarande starka kramper i musklerna och därmed ofta ofrivilliga rörelser eller ryckningar. När Allan - Herndon - Dudley syndrom utvecklas kan de drabbade inte längre tala. Patientens vardagsliv är betydligt begränsad av syndromet och livskvaliteten reduceras. I vissa fall är patienterna sedan beroende av hjälp av andra människor i deras vardag.
Det är vanligtvis inte möjligt att behandla Allan - Herndon - Dudley syndrom orsakssamt. Av denna anledning är behandlingen endast symptomatisk. De drabbade är beroende av olika terapier, som emellertid inte alltid leder till en positiv sjukdomsförlopp. I vissa fall begränsar Allan - Herndon - Dudley syndrom livslängden för de drabbade.
Du hittar din medicin här
➔ Läkemedel mot muskelsvaghetförebyggande
AHDS kan endast förhindras genom genetisk rådgivning. Mutationsbärare kan till exempel besluta att få sina egna barn.
Eftervård
Behovet av uppföljning av genetiskt orsakade Allan - Herndon - Dudley-syndrom drabbar endast manliga spädbarn. Problemet är att det inte finns någon lämplig form av terapi för detta ärftliga tillstånd. De allvarliga konsekvenserna av defekter i sköldkörtelhormonsändare kan knappast förbättras. Försök att ge de drabbade barnen lättnad genom att ge dem speciella sköldkörtelhormoner har misslyckats.
Problemet är att grunden för sjukdomen vanligtvis redan är etablerad i moderns kropp. De skadar det ofödda barnet permanent. Ur denna synvinkel börjar behandlingen för sent, nämligen först efter födseln. I eftervården kan endast de skador som redan finns behandlas. Men det finns hopp. 2014 blev ett fall känt där ett spädbarn med Allan-Herndon-Dudley-syndrom framgångsrikt behandlades med TRIAC. Uppföljning var fortfarande nödvändig eftersom barnet inte kunde botas. Åtminstone lindrade hans symtom.
Allan-Herndon-Dudley-syndrom orsakas delvis av en defekt blod-hjärnbarriär. Detta föreslår studier vid Cedars Sinai Hospital. Sköldkörtelhormonerna kan inte fungera på grund av den defekta blod-hjärnbarriären. Detta gäller även sköldkörtelhormoner som administreras efter att barnet föddes. Bioteknik eller genetisk forskning kan hjälpa till. Alla försök på behandling misslyckas för närvarande. Detta påverkar också uppföljningen av svårt skadade barn.
Du kan göra det själv
Allan-Herndon-Dudley-syndrom är ett allvarligt tillstånd som ännu inte har behandlats effektivt. Men föräldrar kan fortfarande vidta några åtgärder för att stödja terapi.
Först och främst är regelbunden kognitiv träning och fysisk aktivitet viktig. En omfattande terapi, som beroende på svårighetsgraden av syndromet kan bestå av tal- och läsutbildning, men också av allmänna hjärnövningar, erbjuder också övningar från fysioterapi. Utbildningen måste anpassas individuellt till symtomen. Föräldrarna till de drabbade barnen bör därför se till att åtgärderna väljs optimalt och att barnet inte överväldigas.
Vid svåra psykiska störningar kan barnet behöva permanent stöd i vardagen. En öppenvårdstjänst kan vara en viktig lättnad för föräldrarna. Inpatientbehandling är lika viktigt, vilket kan stödjas hemma genom regelbunden övervakning av symtom.
Föräldrar bör också dra fördel av psykologisk rådgivning och vid behov också gå till en självhjälpsgrupp, eftersom kontakten med andra drabbade gör det enklare att hantera sjukdomen. Dessutom får föräldrar ofta viktiga tips om hur man hanterar ett sjukt barn.








.jpg)


.jpg)