Av Niemann-Pick sjukdom är också som Niemann-Pick sjukdom känd. Den ärftliga sjukdomen är en av de lysosomala lagringssjukdomarna.
Vad är Niemann-Pick-sjukdom?

© ktsdesign - stock.adobe.com
Av Niemann-Pick sjukdom är en sjukdom från gruppen av sfingolipidoser. Dessa är metabola sjukdomar som mest manifesterar sig i det centrala nervsystemet. Inom sfingolipidoserna tillhör sjukdomen lysosomala lagringssjukdomar. Dessa kännetecknas av felfunktioner i lysosomerna.
I engelsktalande länder är termen Lysosomala lagringssjukdomar (LSD) används. Vid Niemann-Picks sjukdom deponeras sfingomyelin i levern, benmärgen, mjälten och hjärnan. Sjukdomen fick sitt namn efter upptäckarna Albert Niemann och Ludwig Pick. Det beskrevs först 1914. Niemann-Pick-sjukdomen förekommer ganska sällan.
Runt en nyfödd av 8 000 födslar kommer att utveckla en lysosomal lagringssjukdom. Men det inkluderar inte bara det Niemann-Pick sjukdom, men också sjukdomar som Hunter-syndrom eller Sanfilippo-syndrom.
orsaker
Niemann-Pick-sjukdomen ärvs som ett autosomalt recessivt drag. Vid autosomal recessiv arv är den defekta allelen på en homolog kromosom eller en autosom. Endast homozygota bärare av egenskapen blir sjuka. Detta innebär att det genetiska materialet i en cell måste ha två identiska kopior av den defekta genen på båda kromosomerna för att sjukdomen ska bryta ut.
Niemann-Pick-syndromet är baserat på en genetisk enzymdefekt. Enzymet sfingomyelinas påverkas. Sfingomyelinas ansvarar för klyvningen av sfingomyelin. Enzymdefekten leder till ökad lagring av sfingomyeliner i lysosomerna i mjälten, benmärgen, hjärnan och levern. Lysosomer är cellorganeller som innehåller matsmältningsenzymer.
De smälter främmande material såsom patogener eller cellskräp. De spelar också en viktig roll vid programmerad celldöd (apoptos). I djurförsök kan det visas att uttrycket av den myelingenreglerande faktorn (MRF) reduceras signifikant genom mutationen i NPC-1-genen. Proteinet MRF är en så kallad transkriptionsfaktor. Vid genkodning spelar det en roll i bildandet och skyddet av myelinhöljer.
Myelin-mantlar täcker nervfibrerna och ser till att stimuli överförs snabbt. Antagligen baseras de neurologiska underskotten som uppstår vid Niemann-Picks sjukdom på en felaktig differentiering av oligodendrocyter. Dessa celler tillhör gliacellerna. Deras cellprocesser täcker nervprocessernas cellprocesser som myelinhölje. Således leder den felaktiga differentieringen av oligodendrocyterna till en brist på eller otillräcklig myelinisering.
När det gäller Niemann-Pick typ C-sjukdom försämras också kolesterolmetabolismen. Förutom sfingomyelinerna samlas kolesterol och andra metaboliska produkter också i kroppens celler.
Symtom, åkommor och tecken
Niemann-Picks sjukdom kan delas in i tre former:
- Typ IA är också känd som den akuta infantila neuropatiska formen. Sjukdomen börjar vid tre månaders ålder och manifesteras som en dricksvaghet och utvecklingsstörningar i individuella vävnader och organ.
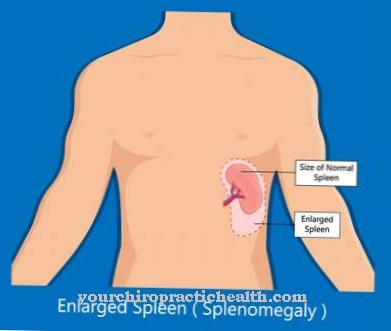
Det huvudsakliga symptomet är svullnad i levern (hepatomegaly). Detta kan också inträffa i kombination med svullnad i mjälten (splenomegaly). Dessutom kan lymfkörtlarna kännas och brun missfärgning av huden inträffar. Neurologisk nedbrytning börjar under andra livet. Påverkade små barn blir döva, blinda och tappar social kontakt.
Prognosen är dålig, vilket innebär att alla barn med Niemann-Pick typ IA-sjukdom kommer att dö inom två år. Denna form är den vanligaste varianten av sjukdomen.
- TYP IS är också känd som den kroniska viscerala formen. Det är en mild kurs med leversvullnad och lunginfiltrat. Det finns inget deltagande i centrala nervsystemet. Patienternas livslängd är endast något begränsad.
- I typ C av Niemann-Picks sjukdom förekommer neonatal gulsot. Hud och sklera hos de drabbade nyfödda färgas gula av avlagringar av färgämnet bilirubin. Supranukleär pares är också typisk för denna variant av sjukdomen. Detta leder till progressiv förlamning av ögonmusklerna med dubbelsyn eller balansstörningar.
Cerebellar ataxi med nedsatt rörelsekoordination kan också observeras. Under sjukdomsförloppet utvecklar patienter ofta sväljstörningar. Detta kan orsaka aspiration lunginflammation. Sjukdomen i typ C är mycket varierande. De första symtomen kan förekomma hos spädbarn, barn eller till och med i tonåren eller i vuxen ålder.
Diagnos & sjukdomsförlopp
Om risken för sjukdomen är känd är diagnosen prenatal möjlig. Om Niemann-Picks sjukdom misstänks, tas vita blodkroppar från benmärgen. Dessa verkar vakuolerade. Detta betyder att leukocyterna har håligheter. Det finns också vakuolerade skumceller.
Detta fenomen kallas "havblå histiocytos". Bristen på aktivitet hos enzymet sfingomyelinas kan detekteras i odlingarna av leukocyter och fibroblaster. Varje andra barn med Niemann-Picks sjukdom visar ett rött makulärt märke under en okulär fundus.
komplikationer
Beroende på typ är Niemann-Pick-sjukdomen associerad med ett antal komplikationer. Med TYP IS kan leversvullnad och lunginfiltrat, dvs ackumulering av främmande kroppar i lungorna. Livsförväntningen för de drabbade är något begränsad och livskvaliteten är ibland allvarligt försämrad. Med typ C kan de första symtomen förekomma i spädbarn.
Detta kan leda till allvarliga utvecklingsstörningar, som ofta är förknippade med cerebellar ataxi med störningar i rörelsekoordination. I samband med sjukdomen uppstår ibland svällande störningar, vilket leder till aspiration lunginflammation och andra komplikationer. De drabbade visar ibland symtom på andnöd, som är förknippat med hosta med sputum, ökad kroppstemperatur och en blå missfärgning av hud och slemhinnor.
I sin tur är sådan cyanos full av allvarliga komplikationer. I TYP IA finns det tidig dricksvaghet och utvecklingsstörningar i organ och vävnader. Leverens svullnad är vanligtvis förknippad med en svullnad i mjälten, vilket orsakar allvarlig fysisk försämring hos de drabbade.
Infektioner förekommer oftare, mag-tarmkanalen blir inflammerad och kroppens egna funktioner minskar snabbt. De drabbade små barnen blir vanligtvis döva och blinda inom två år innan de äntligen dör av de allvarliga komplikationerna av Niemann-Pick-sjukdomen.
När ska du gå till läkaren?
Niemann-Pick-sjukdomen är en ärftlig sjukdom som tar en progressiv kurs. Föräldrar som upptäcker att deras barn har återkommande gulsot och obehag i musklerna bör kontakta barnläkaren. Om det finns motoriska utvecklingsförseningar eller psykologiska beteendestörningar är misstanken om en allvarlig sjukdom som måste diagnostiseras och behandlas uppenbar.
Föräldrar eller vårdnadshavare bör besöka ett specialiserat centrum för sällsynta metaboliska sjukdomar. Barn med Niemann-Pick-syndrom behöver pågående medicinsk behandling på grund av ökande fysiska och psykiska problem.
Ovanliga symtom eller en plötslig ökning av typiska symtom måste rapporteras till den ansvariga läkaren. Detsamma gäller om barnet inte längre kan tolerera den föreskrivna medicinen eller visar andra avvikelser från normalt beteende. Rutinbehandlingar som stopp av medicinering och fysiska undersökningar kan utföras av din husläkare.
De flesta personer med Niemann-Pick-sjukdom måste behandlas av specialister på metabolismsjukdomar. De enskilda symtomen behandlas av neurologer, ortopeder och logopeder. Dessutom är fysioterapeuter och arbetsterapeuter involverade i behandlingen. En terapeut kan också kallas för psykologiska klagomål som depression eller illusioner. På grund av det stora antalet möjliga symtom måste Niemann-Pick-sjukdomen vanligtvis behandlas av ett team av läkare.
Terapi och behandling
En kausal terapi är för närvarande inte känd. Det finns dock bevis för att speciella cyklodextriner kan lindra symtomen på sjukdomen. Cyklodextriner är cykliska oligosackarider som ofta används som lösningsmedel vid läkemedelsproduktion. Niemann-Pick typ C-sjukdom behandlas med miglustat.
Miglustat är ett läkemedel som endast är godkänt i Europeiska unionen för behandling av Niemann-Pick-sjukdomen och för behandling av Gauchers sjukdom typ 1. Läkemedlet är ett iminosugar och ett n-butylderivat av moranolin.
Du hittar din medicin här
➔ Läkemedel mot smärtaOutlook & prognos
Prognosen för Niemann-Pick-sjukdomen är dålig. Sjukdomen är en genetisk defekt. Nuvarande lagstiftning förbjuder forskare att interferera med eller modifiera mänsklig genetik. Även om sjukdomen kan diagnostiseras före födseln, är inget botemedel möjligt baserat på lagkrav.
Fram till idag har läkare och medicinska specialister koncentrerat sig på att utveckla tillräcklig medicinsk vård efter att personen är född. Behandlingen består för närvarande av att initiera läkemedelsbehandling för att stödja patientens ämnesomsättning så bra som möjligt. Som ett resultat är optimeringar redan möjliga i patientens utvecklingsprocess, vilket bidrar till att förbättra den totala situationen.
Utan behandling reduceras den drabbade personens livskvalitet kraftigt. Dessutom kan livshotande tillstånd utvecklas, eftersom sjukdomen åtföljs av svullnad av inre organ och andnöd. Risken för en akut situation ökar avsevärt utan behandling. Långtidsbehandling indikeras därför oavsett intensiteten hos de enskilda symptomen. Patienterna behöver daglig vård och stöd för att hantera vardagen. Beroende på vilken typ av sjukdom som förekommer kan patienten dö för tidigt under de första åren av livet om sjukdomen utvecklas dåligt.
förebyggande
Niemann-Picks sjukdom ärvs som ett autosomalt recessivt drag. Det finns för närvarande inget effektivt förebyggande.
Eftervård
I de flesta fall har den drabbade personen endast några få och begränsade uppföljningsåtgärder tillgängliga för Niemann-Pick-sjukdomen. Av detta skäl måste patienten konsultera en läkare vid de första tecknen och symtomen så att det inte finns andra komplikationer eller klagomål. Ju tidigare en läkare kontaktas, desto bättre desto längre är sjukdomen, så att en läkare bör konsulteras så snart de första symtomen eller tecknen visas.
Om patienten vill få barn, bör genetisk testning och rådgivning definitivt utföras för att förhindra återfall av Niemann-Pick-sjukdomen. De flesta av patienterna är vanligtvis beroende av intaget av olika läkemedel.
Den drabbade ska alltid uppmärksamma rätt dosering och även regelbundet intag för att permanent lindra symtomen. Om något är oklart eller om du har några frågor bör en läkare alltid konsulteras först. På samma sätt är många av patienterna beroende av hjälp och stöd från sina egna familjer i deras vardag. Framför allt kan depression och andra psykologiska klagomål lindras.
Du kan göra det själv
Möjligheterna till självhjälp är extremt begränsade med Niemann-Pick-sjukdomen. Speciellt typ IA erbjuder inte tillräckliga möjligheter att förbättra situationen. Det sjuka barnets förväntade livslängd är mycket låg trots alla ansträngningar.
I vardagen bör fokus därför ligga på att göra tiden tillsammans så trevlig som möjligt. Att njuta av fritiden är viktigt för att bygga närhet, solidaritet och stabilitet. Sjukdomen är en enorm utmaning för både patienter och släktingar. Att bygga upp mentala krafter är särskilt viktigt när man hanterar motgångar. Av detta skäl är psykologiskt stöd viktigt för alla inblandade.
För många är det också en hjälp om det finns möjlighet till utbyte med andra drabbade personer. Det kan därför vara fördelaktigt att kontakta etablerade självhjälpsgrupper. I gemensamma diskussioner sker ett utbyte på grundval av ömsesidig förståelse. Kommunikation kan hjälpa till med bearbetning. Det ger också tips för att hantera väl.
Mental teknik och avslappningsövningar främjar minskning av stressfaktorer. Eftersom situationer med överdriven stress och därmed vegetativa problem uppstår ofta, kan träningsenheterna bidra till att minska stress. Hanteringen av den övergripande situationen bör därmed förbättras.
.jpg)
.jpg)
.jpg)
.jpg)


.jpg)