Det finns 45 olika totalt lysosomala lagringssjukdomar, som är en heterogen grupp medfödda metaboliska sjukdomar. Människor som lider av någon av dessa sjukdomar har en genetisk defekt. Alla lagringssjukdomar har en sak gemensamt: ett visst enzym är antingen frånvarande eller endast delvis funktionellt.
Vad är lysosomal lagringssjukdom?

© designua - stock.adobe.com
Dessa medfödda lagringssjukdomar är sällsynta, eftersom färre än fem av 10 000 personer drabbas. De olika sjukdomarna har en mycket annorlunda kurs, och symtomen kan variera mycket.
De mest kända formerna av lysosomal lagringssjukdom är Fabrys sjukdom, Gauchers sjukdom, Pompesjukdom och mukopolysackaridos (MPS). De kallas ofta för "föräldralösa barn" eftersom vägen till en specifik diagnos och lämplig terapi kan vara mycket lång. Ibland kan det ta år för de drabbade att ta reda på vad som händer dem.
orsaker
Lysosomala lagringssjukdomar kännetecknas av vissa former av ärftliga metaboliska sjukdomar. Patienterna saknar ett viktigt enzym som säkerställer att metabolismbalansen går smidigt. I den mindre uttalade formen är detta enzym åtminstone inte närvarande i tillräckliga mängder.
Enzyms uppgift är att avyttra föroreningar och avfallsämnen som ackumuleras i den mänskliga organismen via metabolismen via lysosomerna, eller att bearbeta dem igen på ett sådant sätt att symptom inte uppstår.
Om det finns en enzymbrist, garanteras inte längre denna väl fungerande bortskaffningscykel. De skadliga ämnena sätter sig i cellerna och stör metabolismcykeln. I den inledande fasen har störningarna inte någon märkbar effekt, det finns bara några få begränsningar. Men om denna metabola störning förblir obehandlad till följd av en enzymbrist multipliceras symtomen eftersom cellerna blir mycket förstorade.
Symtom, åkommor och tecken
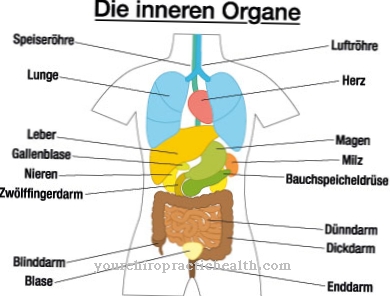
I värsta fall går dessa under. Konsekvenserna är skador på ben, nervsystemet, mjälte, njurar, muskler eller hjärta. Fabrysjukdom gör att fett (globotriaosylceramid, Gb3) lagras i cellerna på grund av den minskade eller frånvarande enzymaktiviteten. Dessa oönskade avlagringar kan leda till svår smärta i tår eller fingrar, stroke och njurskador.
Diagnos & sjukdomsförlopp
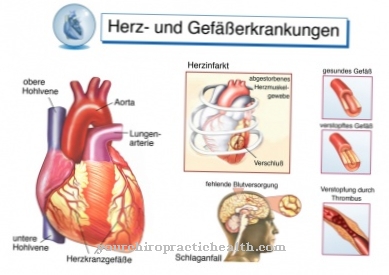
Denna kliniska bild påverkar olika system samtidigt: blodkärl, njurar, hjärta och nervsystem. Den autosomala recessiva ärvda Gauchersjukdomen orsakar en mutation av enzymet "beta-glukocerebrosidas" och leder till en ansamling av substrat i cellerna, speciellt i makrofagerna (scavenger celler), som tillhör retikulo-endotelialsystemet. Blodantalet förändras, levern och mjälten förstoras och benen skadas.
Sjukdomen är progressiv och är mestadels etnisk, eftersom den i de flesta fall förekommer hos människor av judisk härkomst. Pompesjukdom är också känd som "syra maltasbrist". Den kliniska bilden tillhör gruppen glykogenes typ II. De drabbade personerna saknar enzymet "alfa-1,4-glukosidas" (surt maltas) eller är inte tillgängligt i tillräckliga mängder. På grund av nedsatt glykogennedbrytning i musklerna lider patienter av förstörelsen av muskelcellerna i form av sockerlagring.
Mucopolysaccharidosis typ I (MPS), även känd som Jagers sjukdom, har olika kliniska orsaker. Hurlers sjukdom är den svåraste formen och Scheies sjukdom är i slutet av den kliniska patogenesen. Det finns övergångar med olika egenskaper mellan dessa två former av progression. Den mest slående egenskapen är den nedsatta nedbrytningen av kolhydrater som samlas i cellernas lysosomer.
Jagaresjukdomspatienter kan uppleva kort statur, förstorad mjälte och lever, grova egenskaper, förtjockad hud, förstorad tunga och andningssvårigheter Dessutom förändras skelettet ofta i området med bäcken, ryggraden, handbenen och skallen. Umbilical och [[inguinal hernias] är möjliga.
komplikationer
I de flesta fall förekommer symtom eller komplikationer mycket sent vid denna sjukdom. På grund av detta diagnostiseras det sent, vilket gör att tidig behandling i de flesta fall är omöjlig. Utan behandling uppstår olika klagomål och skador på de inre organen när sjukdomen utvecklas.
Njurarna, levern och mjälten påverkas särskilt. Hjärtat kan också påverkas av denna sjukdom, vilket i värsta fall kan leda till hjärtdöd. Dessutom uppstår skador på njurarna och de drabbade lider ofta av smärta i tår eller fingrar. Förlamning kan också uppstå om hjärnan har skadats av denna sjukdom. Lever och mjälte kan förstoras och orsaka allvarlig smärta också.
Det är inte ovanligt att den drabbade personens ben är spröda och smärtsamma. Behandling av denna sjukdom visar sig vara svår. I många fall minskas livslängden för den drabbade avsevärt. Det finns vanligtvis inga speciella komplikationer när du använder medicinering. En positiv förlopp av sjukdomen kan dock inte garanteras i alla fall.
Du hittar din medicin här
➔ Läkemedel mot smärtaNär ska du gå till läkaren?
Hårförlust, ledproblem och organsjukdomar är möjliga tecken på lysosomal lagringssjukdom. Ett läkarbesök rekommenderas om symtomen fortsätter att återkomma eller om de plötsligt uppträder utan att en orsak hittas. Om symtomen är relaterade till en redan diagnostiserad enzymdefekt eller annan allvarlig sjukdom, bör den ansvariga läkaren konsulteras. En obehandlad lagringssjukdom kan leda till demens, infertilitet, neuropatier och andra komplikationer, av vilka några är livshotande. Därför bör alla tänkbara symtom undersökas, även om det inte finns någon specifik misstank.
Symtomen på lysosomal lagringssjukdom kan förekomma i faser eller utvecklas lumskt, men kräver alltid undersökning och behandling. Berörda människor är bäst att prata direkt med sin husläkare eller en internist. Den faktiska behandlingen sker vanligtvis i en specialistklinik för interna sjukdomar, även om fysioterapi eller psykoterapi kan anslutas beroende på symtom. I synnerhet indikeras terapeutiska åtgärder på grund av den ofta negativa sjukdomsförloppet.
Terapi och behandling
Beroende på hur tidigt en adekvat diagnos ställs, kan dessa ärftliga sjukdomar behandlas mycket bra med enzymersättningsterapi, så att de drabbade har mycket färre symtom och därmed en bättre livskvalitet. Denna ersättningsterapi används enligt den kliniska bilden.
Personer som lider av Gauchers sjukdom saknar ”enzymet ß-glukocerebrosidas”, som produceras bioteknologiskt och infunderas i patientens kropp. Lysosomer verkar effektivt och kan absorbera ämnen från deras omedelbara miljö. Av detta skäl modifieras de artificiellt använda enzymerna på ett sådant sätt att de kan tillföras lysosomerna på ett idealt sätt.
Makrofagerna (fagocyter) bryter ner glukocerebrosiderna som samlats i cellerna. Denna terapi kan jämföras med insulinbehandling för diabetes mellitus, med skillnaden att det inte är ett saknat hormon, utan ett icke-existerande enzym som tillhandahålls. Kroppen bryter regelbundet ner alla ämnen, inklusive det medföljande konstgjorda enzymet.
På grund av denna regelbundna nedbrytning av ämnet måste patienterna genomgå denna infusionsterapi regelbundet till slutet av sitt liv. Enzymersättningsterapin verkar inte symptomatiskt utan bekämpar direkt orsaken till den ärftliga sjukdomen. Läkare kallar denna terapi kausal. Terapiprinciperna ska användas för alla fyra av de ovan nämnda vanliga lagringssjukdomarna.
Pompe-patienter behandlas också med infusionsterapi. Vid denna sjukdom tillhandahålls det icke-existerande enzymet "syra alfa glukosidas" och hjälper till att bryta ned glykogen som har samlats i musklernas lysosomer. Hos patienter med sjukdomstypen "mukopolysackaridos typ I" är det lysosomala enzymet "alfa-iduronidas" inte närvarande eller inte närvarande i tillräckliga mängder. Det är en av de sällsynta lagringssjukdomarna där sockermolekyler samlas i organ och vävnader.
Om processen är normal, bryter enzymet upp mukopolysackarider. Sockermolekylerna är långkedjiga och är involverade i utvecklingen av stöd- och bindväv, till exempel ben, hud, ledvätskor och brosk. Om den normala nedbrytningsförloppet störs på grund av bristen på enzym, samlas patologiska glykosaminoglykaner (GAG) i de enskilda cellerna. Framtida behandlingsalternativ syftar till att ta tabletter.
Outlook & prognos
Prognosen för lagringssjukdom är dålig. En genetisk disposition visade sig vara orsaken till hälsoproblemen. Lagliga krav förbjuder läkare och forskare att ändra mänsklig genetik. Av denna anledning förblir sjukdomen livslångt och har inga möjligheter att återhämta sig.
Den behandlande läkaren koncentrerar sig på att behandla symtomen som uppstår. Om de inte behandlas ökar olika klagomål med tiden. Bensystemet är skadat och problem med organen uppstår. I värsta fall kommer det att finnas funktionella svårigheter i de inre organen och i slutändan ett fel i organaktiviteten. Detta hotar den person som berörs av för tidig död.
Utmaningen med sjukdomen ligger i diagnosen. Hos ett stort antal patienter uppträder signifikanta och starkt märkbara symtom först senare i livet. Som ett resultat förblir den genetiska störningen obemärkt under lång tid och tidig behandling av sjukdomen är svår. Ju senare en diagnos ställs, desto ogynnsammare är den fortsatta kursen. I ett avancerat stadium av sjukdomen är de inre organen eller lederna redan allvarligt skadade. Kirurgiska ingrepp krävs och om sjukdomen utvecklas ogynnsamt kan bara ett givarorgan rädda den drabbade personens liv. Tidig behandling är därför viktigt för en förbättrad prognos.
förebyggande
Eftersom det är en medfödd genetisk defekt som förhindrar uttryck av ett enzym, kan denna sjukdom inte behandlas förebyggande. De senaste prestationerna inom genteknik kan emellertid ge en strategi på detta område.
Eftervård
Med denna sjukdom lider människor av ett antal olika komplikationer och sjukdomar. Som regel har alla en mycket negativ effekt på livskvaliteten för den drabbade, så att en diagnos bör ställas mycket tidigt. Ju tidigare en läkare konsulteras, desto bättre är den fortsatta sjukdomen vanligtvis.
Svårighetsgraden av denna sjukdom kan vara mycket olika, så att en allmän förutsägelse ofta inte är möjlig. De drabbade drabbas av allvarliga skador på de inre organen. Njurarna och hjärtat påverkas främst så att barnet kan dö under de första dagarna om symptomen inte korrigeras i tid. Det finns också fettavlagringar i olika delar av kroppen.
Fingrar och tår är särskilt drabbade, vilket kan leda till avsevärt minskad estetik för den drabbade. Som regel skadas njurarna och hjärnan vidare, så att den berörda personen dör till följd av denna skada. Föräldrar och släktingar lider också ofta av depression eller andra psykiska störningar på grund av sjukdomen.
Du kan göra det själv
Lysosomala lagringssjukdomar kräver ofta intensiv medicinsk vård. Ofta finns det inte tillräckligt med möjligheter till självhjälp. Föräldrarna till drabbade barn upplever ofta svår stress i sin hemmiljö eftersom deras barn behöver ständig vård och uppmärksamhet.
De kliniska bilderna av de individuella lagringssjukdomarna är olika. Det finns både enkla och mycket svåra former. Ett exempel är Gauchers sjukdom. Föräldrarnas hjälp är ofta begränsad till att mata det svårt funktionshindrade barnet. I mildare fall kan livslängden vara nästan normal. Icke desto mindre är ständig medicinsk övervakning nödvändig för att avvärja möjliga komplikationer. Regelbunden fysisk aktivitet är en av de medföljande terapierna som också kan utföras hemma. Dessutom måste en noggrann cancerscreeningsundersökning ordnas. Det kräver ständiga besök hos läkaren med sina barn från föräldrarna. Detsamma gäller för andra lysosomala lagringssjukdomar.
Vid vissa sjukdomar, utöver fysiska funktionsnedsättningar, kan också mentala begränsningar uppstå, som fortfarande kräver särskilt stöd. I mildare former av vissa sjukdomar, till exempel Jagers sjukdom, uppstår initialt bara skelettförändringar och ansiktsdysmorfismer. Här kan dock den drabbade patienten ofta leva ett självständigt liv. Men här krävs också ständiga medicinska undersökningar för att utesluta möjliga komplikationer såsom hjärtsvikt eller luftvägssjukdomar. Patienten kan hantera psykologisk stress orsakad av fysiska deformationer genom psykologisk rådgivning.
.jpg)

.jpg)

.jpg)