Lysyloxidas är ett enzym av bindväv som har katalytiska uppgifter och främjar tvärbindning av kollagen och elastin. Enzymet har en stabiliserande effekt på bindvävnaden genom att utföra oxidativ deamination och därmed skapa de grundläggande förutsättningarna för tvärbindning. I Cutis laxa reduceras aktiviteten för lysyloxidas.
Vad är lysyloxidas?
Det finns olika enzymer i människokroppen, som alla har katalytisk aktivitet. Enzymer möjliggör reaktioner i människokroppen eller påskyndar dem. Lysyloxidas är ett enzym som finns i humant bindväv. Det kallas också proteinlysin 6-oxidas och finns främst i det extracellulära utrymmet i bindvävnaden.
Den katalytiska aktiviteten hos enzymet avser i detta fall tvärbindningen mellan kollagen och elastin. Lysyloxidas stabiliserar de två proteinerna på ett mekaniskt sätt och möjliggör således den reaktiva anslutningen. Lysyloxidas finns inte bara i människokroppen. Andra ryggradsdjur är också utrustade med enzymet. Lysyloxidas anses vara en stabilisator av bindvävnaden. En brist i enzymet leder till den kliniska bilden av cutis laxa, en allvarlig och ärftlig svaghet i bindvävnaden.
Funktion, effekt och uppgifter
Lysyloxidas tar viktiga uppgifter i det extracellulära utrymmet i tvärförbindelsen mellan enskilda kollagenmolekyler. I människokroppen spelar kollagen en viktig roll inom proteinerna, med cirka 30 procent av den totala proteinmassan.
Kollagen är det vanligaste proteinet. Det är ett strukturellt och byggande protein som utgör många komponenter i kroppen, såsom bindväv, ben, tänder, brosk, senor, ligament och hud. Lysyloxidas stödjer bindningen av kollagen till karbonylgrupper och bidrar således till stabiliteten hos de nämnda kroppskomponenterna. Den har katalytisk aktivitet för framställning av karbonylgrupper som bildar kovalenta tvärbindningar på kollagener i aldolkondensationer. Den katalytiska uppgiften för lysyloxidas är därför att förbereda sig för fibrilbildning. Enzymet skapar alla kemiska tillstånd som är nödvändiga för bildning.
Fibriller betraktas som fibrer av fiber. De motsvarar tunna och fibrösa delar av kroppen och finns i växtcellväggar, i mänskliga muskler och i bindväv. Uppgiften för lysyloxidas i detta sammanhang är väsentligen oxidativ deaminering av lysylrester. I kemi är deamination den kemiska uppdelningen av aminogrupper som ammoniumjoner eller ammoniak. Oxidativ deamination splittrar aminogrupper av aminosyran L-glutamat från väte och oxiderar dem till iminogrupper med överföring av väte till NAD + eller NADP +.
Detta följs av hydrolytisk klyvning av iminogrupper som ammoniumjoner, vilket är associerat med bildningen av a-ketosyra. Deamination motsvarar det första steget i den biokemiska nedbrytningen av aminosyror, som i däggdjur huvudsakligen äger rum i levern. Ammoniumjonen som bildas under deaminering omvandlas till urea. Deamineringsprocesserna för lysyloxidas ger upphov till aldehydgrupper som med de enskilda aminogrupperna i andra lysylrester skapar så kallade Schiff-baser och på detta sätt kan bilda de stabiliserande tvärbindningarna i kollagen.
Utbildning, förekomst, egenskaper och optimala värden
Lysyloxidas i DNA kodas av LOX-genen, som hos människor är belägen på kromosom 5 i genlokus q23.3 till q31.2. Genprodukten är inte den slutliga formen av enzymet. Produkten är inte ett färdig lysyloxidas, utan en föregångare som efter translation har en molmassa på 47 kDa.
Glykosylering sker i den fortsatta kursen. Under denna process ökar den senare enzymets molmassa till 50 kDa och föregångarformen av lysyloxidas utsöndras i det extracellulära utrymmet. Efter utsöndring bearbetas pre-pro-lysyloxidaset vidare. Ämnet är uppdelat i det extracellulära utrymmet. Protein 1 ansvarar för uppdelning i två fragment, på det här sättet å ena sidan produceras 32 kDa lysyloxidas. Å andra sidan skapas en restsubstans, som i detta fall motsvarar en polypeptid.
Sjukdomar och störningar
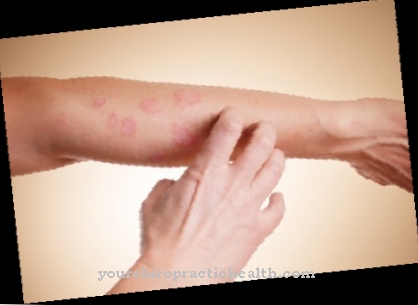
Genetiska defekter i lysyloxidas kan orsaka den kliniska bilden av cutixlax. Denna sjukdom kallas också dermatokalas och avser en grupp ofta åldersrelaterade svagheter i bindvävnaden, som i de flesta fall observeras med familjär ansamling.
Det vanliga kännetecknet för alla dermatokalasfenomen är slak och oelastisk hud, som ofta hänger ner i stora veck på olika delar av kroppen. De flesta av de drabbade ser äldre ut än de är på grund av förändringarna. Sjukdomen orsakas bland annat av genetiska mutationer. I detta sammanhang talar vi om cutis laxa-syndrom. Sjukdomen kan existera i autosomala recessiva, autosomalt dominerande och x-kromomala former. I många fall är cutis laxa-syndromet förknippat med andra avvikelser och, om organen är involverade, till exempel, kan det vara dödligt.
ARCL1 motsvarar en cutis laxa av den autosomala recessiva typen 1 och anses vara den svåraste formen, som under vissa omständigheter kan leda till livshotande komplikationer. Formen ARCL1A är associerad med mutationer i FBLN5-genen på locus 14q32.12. Typ ARCL1B är associerad med mutationer i EFEMP2-genen på locus 11q13.1 och variant ARCL1C motsvarar en cutis laxa med tillhörande anomalier i lunga, mag-tarmkanalen och urinvägarna, som beror på mutationer i LTBP4-genen på lokus 19q13.2.
Mutationerna i de nämnda generna leder till en aktivitet under lyxloxidas under genomsnittet. Otillräckliga tvärförbindelser skapas på grund av enzymets minskade aktivitet. Patientens bindväv stabiliseras inte tillräckligt.



.jpg)